Computational Studies of Molecular Bismuth Complexes
In progress
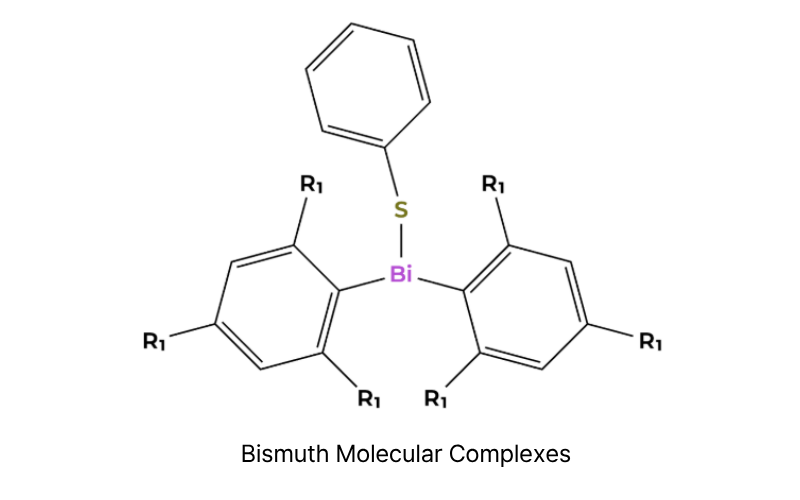
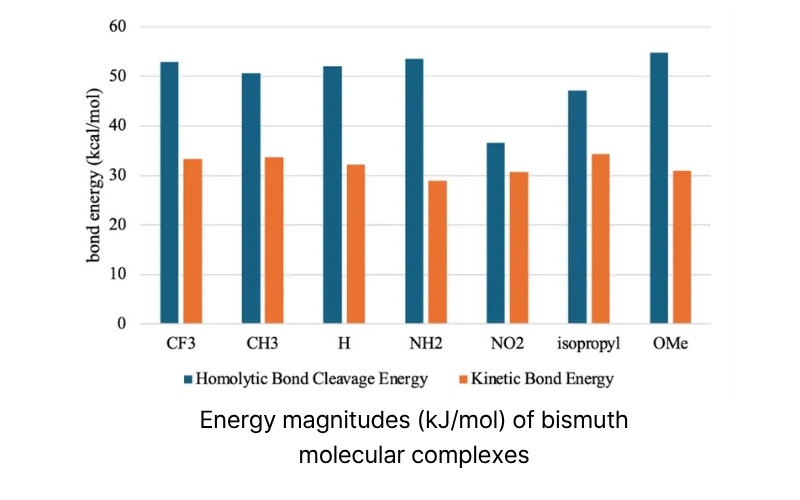
Abstract: Computational analysis of effects of ligand architecture on bond strength and radical stability in molecular bismuth complexes through using Density Functional Theory (DFT) and Quasi-Atomic Orbital (QUAO)
Methods: The bond dissociation energy (BDE) calculations through ground-state geometry optimization and vibrational frequency analysis via DFT (M06-L/def2-TZVP/ECP), followed by QUAO analysis of electronic structure (σ bonds, delocalized π orbitals, hyperconjugation, π backbonding)
Implication: Advancing sustainable main-group catalysis through predictive electronic structure modeling of ligand architectures to replace transition-metal catalysts