Background
Transition metals are commonly used in chemical reactions because they can easily gain and lose electrons, helping form and break chemical bonds. However, many of these metals—like palladium or platinum—are expensive, toxic, and harmful to the environment. This has led scientists to look for safer and more sustainable alternatives. One promising option is bismuth, a main-group element that is much less toxic and more affordable but can still take part in similar types of reactions. In recent years, researchers have discovered that with the right ligands—the molecules attached to the metal—bismuth compounds can form and break bonds in ways similar to traditional transition-metal catalysts. These ligands are important because their size and electronic properties can help stabilize the reactive parts of the molecule and control how easily bonds break. Yet, it is still unclear how changes in ligand design affect the strength of bismuth bonds and the behavior of the resulting radicals.
This study uses computer modeling to explore how different ligand structures influence bond breaking and electron distribution in molecular bismuth compounds, with the goal of guiding the design of safer and more efficient catalysts.
Methods
- DFT: DFT: M06L/def2-TZVP with CPCM
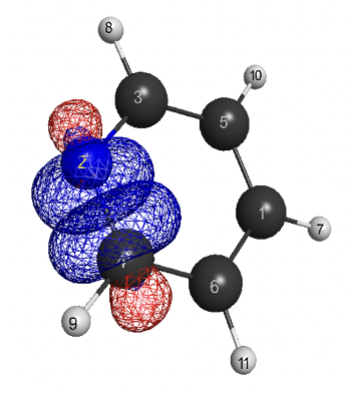
- Orbital Descriptors: QUAO to probe π-acceptor/σ-donor effects
- Substituent Series: –F, –OCH3, –CH3, –NO2, –NH2, etc.
- Bridging Group Series: Sulfonyl, Methyl, Sulfur
Key Results(in progress)
- Early trend: stronger π-acceptor ligands lower effective Bi–H cleavage barrier
Implication
Establishing ligand design rules to enable stable, reactive bismuth catalysts as sustainable alternatives to precious transition metals
Quantitative QUAO analysis with clear correlations between ligand donor strength and Bi–X bond dissociation energies
Docs